- HOME
- Enzyme List
- GTD-209・309 GLUTAMATE DEHYDROGENASE (NADP-dependent)
-
GTD-209・309
GLUTAMATE DEHYDROGENASE (NADP-dependent) from Proteus sp.

PREPARATION and SPECIFICATION
| Appearance | Solution with 50 mM Tris-HCl buffer containing 0.05 % NaN3 and 5.0 mM EDTA, pH 7.8 | |
|---|---|---|
| Activity | GradeⅡ・Ⅲ 300 U/mg-protein or more (9,000U/mL or more) |
|
| Contaminants | NADPH oxidase | ≤ 1.0×10-2 % |
| Glutathione reductase | ≤ 1.0×10-2 %(GradeⅡ-209) | |
| ≤ 1.0×10-1 %(GradeⅢ-309) | ||
| Stabilizer | Ethylenediaminetetraacetic acid (EDTA) | |
PROPERTIES
| Stability | Stable at 4 ℃ for at least one year(Fig.1,2) |
|---|---|
| Molecular weight | approx. 300,000 |
| Isoelectric point | 4.6 |
| Michaelis constants | 1.1×10-3 M (NH3), 3.4×10-4 M(α-Ketoglutarate) 1.2×10-3 M (L-Glutamate), 1.4×10-5 M(NADPH), 1.5×10-5M(NADP+) |
| Structure | 6 subunits (M.W.50,000) per enzyme molecule |
| Inhibitors | Hg2+, Cd2+, p-chloromercuribenzoate, pyridine, 4-4'-dithiopyridine, 2,2'-dithiopyridine |
| Optimum pH | 8.5 (α-KG→L-Glu) 9.8 (L-Glu→α-KG)(Fig.5) |
| Optimum temperature | 45 ℃ (α-KG→L-Glu) 45−55 ℃ (L-Glu→α-KG)(Fig.6) |
| pH Stability | pH 6.0−8.5 (25 ℃, 20 hr)(Fig.7) |
| Thermal stability | below 50 ℃ (pH 7.4, 10 min)(Fig.8) |
| Substrate specificity | (Table 1) |
APPLICATIONS
This enzyme is useful for enzymatic measurement of NH3, α-ketoglutaric acid, and L-glutamic acid, and for assays of leucine aminopeptidase and urease. It is also used for enzymatic measurement of urea in combination with urease (URH-201), in clinical analysis.
ASSAY
Principle

The formation of NADPH is measured at 340nm by spectrophotometry.
Unit definition
One unit causes the oxidation of one micromole of NADPH per minute under the conditions detailed below.
Method
Reagents
| A. Buffer solution | 0.1 M Tris-HCl buffer, pH 8.3 | |
|---|---|---|
| B. NH4Cl solution | 3.3 M | |
| C. α-Ketoglutarate solution | 0.225 M (adjust the pH to 7.0−9.0 with NaOH)(Should be prepared fresh) | |
| D. NADPH solution | 7.5 mM (Should be prepared fresh) | |
| E. Enzyme diluent | 50 mM K-Phosphate buffer, pH 6.6 containing 0.2 % BSA and 50mM EDTA | |
Procedure
1. Prepare the following reaction mixture in a cuvette (d = 1.0cm) and equilibrate at 30 ℃ for approximately 5 minutes.
| 2.5 mL | Buffer solution | (A) |
| 0.2 mL | NH4Cl solution | (B) |
| 0.1 mL | α-Ketoglutarate solution | (C) |
| 0.1 mL | NADPH solution | (D) |
| Concentration in assay mixture | |
|---|---|
| Tris-HCl buffer | 85 mM |
| α-Ketoglutarate | 7.6 mM |
| NH4Cl | 0.22 M |
| NADPH | 0.25 mM |
| EDTA | 0.85 mM |
2. Add 0.05 mL of the enzyme solution* and mix by gentle inversion.
3. Record the decrease in optical density at 340 nm against water for 2 to 3 minutes with a spectrophotometer thermostated at 30 ℃ and calculate the ΔOD per minute from the linear portion of the curve (ΔOD test).
At the same time, measure the blank rate (ΔOD blank) using the same method as the test except that the enzyme diluent (E) is added instead of the enzyme solution.
*Dilute the enzyme preparation to 0.4−0.9 U/mL with ice-cold enzyme diluent (E), immediately before the assay.
Calculation
Activity can be calculated by using the following formula :
Volume activity (U/mL) =
-
ΔOD/min (ΔOD test−ΔOD blank)×Vt×df
6.22×1.0×Vs
= ΔOD/min×9.486×df
| Vt | : Total volume (2.95 mL) |
| Vs | : Sample volume (0.05 mL) |
| 6.22 | : Millimolar extinction coefficient of NADPH (cm2/micromole) |
| 1.0 | : Light path length (cm) |
| df | : Dilution factor |
REFERENCES
1) H.Shimizu, T.Kuratsu and F.Hirata; J.Ferment.Technol., 57, 428 (1979).
Table 1. Substrate Specificity of Glutamate dehydrogenase
-
Substrate (50mM) Relative activity(%) L-Glutamate 100 L-Norvaline 0.39 L-α-Aminobutyrate 0.19 L-Norleucine 0.04 D,L-Homocysteine 0.03 L-Isoleucine 0.02 -
Substrate (50mM) Relative activity(%) L-Glutamine < 0.01 L-Aspartate < 0.01 L-Asparagine < 0.01 L-Valine < 0.01 L-Leucine < 0.01 L-Alanine < 0.01 L-Methionine < 0.01
Glutamate dehydrogenase:18 U/mL of 0.1M glycine-NaOH buffer, pH 9.0 NADP+: 0.3 mM
-
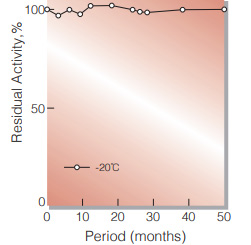
Fig.1. Stability (GTD-209) (Solution)
50 % glycerol solution in 25mM Tris-HCI buffer contg. 2.5mM EDTA, pH7.8 enyzme concentration: 5,000U/mL
-
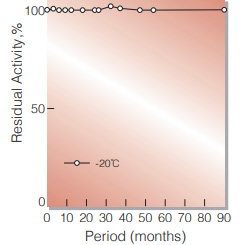
Fig.2. Stability (GTD-309) (Solution)
50 % glycerol solution in 25mM Tris-HCI buffer contg. 2.5mM EDTA, pH7.8 enyzme concentration: 5,000U/mL
-
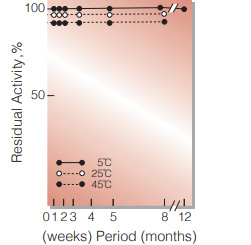
Fig.3. Stability (Solution)
50 % glycerol solution in 25mM Tris-HCI buffer contg. 2.5mM EDTA, pH7.8 enyzme concentration: 5,000U/mL
-
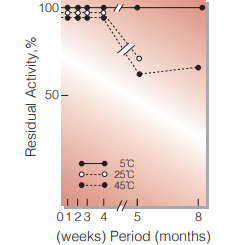
Fig.4. Stability (Suspension)
3.0M ammonium sulfate suspension in 50mM Tris-HCI buffer containing 5mM EDTA. pH7.8 enzyme concentration : 10,000U/mL
-
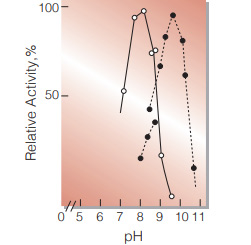
Fig.5. pH-Activity
○̶̶○,α-KG →L-Glu;●̶̶●,L-Glu→α-KG in 0.1M buffer solution:pH7.4-8.8, Tris-HCI;pH8.7-10.7,glycine-NaOH
-
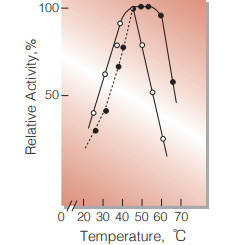
Fig.6. Temperature activity
○̶̶○,α-KG→L-Glu: 0.1M Tris-HCI buffer, pH8.3;●̶̶● , L-Glu→α-KG:0.1M glycineNaOH buffer,pH10.0
-
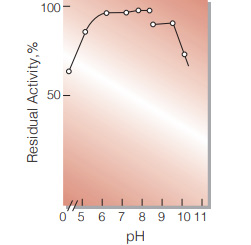
Fig.7. pH-Stability
25℃, 20hr-treatment with 0.1M buffer solution: pH4.4-6.2, acetate; pH6.2-8.4, phosphate; pH8.8-10.2, glycine-NaOH
-
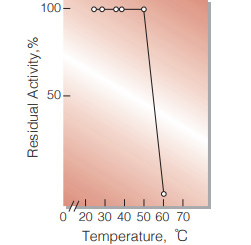
Fig.8. Thermal stability
10min-treatment with 0.1M K-phosphate buffer,pH7.4
活性測定法(Japanese)
1. 原理
NADPHの消失量を340nmの吸光度の変化で測定する。
2.定義
下記条件下で1分間に1マイクロモルのNADPHが酸化される酵素量を1単位(U)とする。
3.試薬
- 0.1M Tris-HCl緩衝液, pH8.3
- 3.3M NH4Cl水溶液
- 0.225M α-ケトグルタル酸水溶液(NaOHでpHを7.0〜9.0に調整)(用時調製)
- 7.5mM NADPH水溶液(用時調製)
酵素溶液:分析直前に酵素標品を予め氷冷した0.2 %BSAと50mM EDTAを含む50mM Kリン酸緩衝液, pH6.6で0.4〜0.9U/mlに希釈する。
4.手順
1.下記反応混液をキュベット(d=1.0cm)に調製し,30℃で約5分間予備加温する。
| 2.5 mL | Tris-HCl緩衝液 | (A) |
| 0.2 mL | NH4Cl水溶液 | (B) |
| 0.1 mL | α-ケトグルタル酸水溶液 | (C) |
| 0.1 mL | NADPH水溶液 | (D) |
2.酵素溶液を0.05 mLを添加し,ゆるやかに混和後,水を対照に30℃に制御された分光光度計で340nmの吸光度変化を2〜3分間記録し,その初期直線部分から1分間当りの吸光度変化を求める(ΔOD test)。
3.盲検は反応混液①に酵素溶液の代りに酵素希釈液(0.2 %BSAと50mM EDTAを含むK-リン酸緩衝液,pH6.6)を加え,上記同様に操作を行って,1分間当りの吸光度変化を求める(ΔODblank)。
5.計算式
U/mL =
-
ΔOD/min (ΔOD test−ΔOD blank)×2.95(mL)×希釈倍率
6.22×1.0×0.05(mL)
| = ΔOD/min×9.486×希釈倍率 | |
| 6.22 | : NADHのミリモル分子吸光係数(cm2/micromole) |
| 1.0 | : 光路長(cm) |
CONTACT
-
For inquiries and cosultations regarding our products, please contact us through this number.
- HEAD OFFICE+81-6-6348-3843
- Inquiry / Opinion